
Cleanroom validation plays a crucial role when bringing a cleanroom online. It must be done thoroughly and accurately to achieve a suitable environment for its intended use and to meet applicable regulatory requirements. Companies should expect auditors to review the documentation that is generated from this validation, so it is imperative that all data is accurate, aligned to cGMP standards, and managed and retained in an organised manner.
The team at Analytical Lab Group believes cleanroom validation is a critical aspect and should be a high priority at your company to protect the quality of your product.
Before their initial use, cleanrooms need to be validated by a qualified team to ensure they are functioning properly. This includes ensuring they are capable of meeting the International Organization for Standardization (ISO) specifications for those areas and can maintain the required environment when the area is released for use.
The three major components of a cleanroom validation are installation qualification (IQ); operational qualification (OQ); and performance qualification (PQ).
Each step can be described as follows:
- The IQ serves as a test plan to verify that all the critical installation parameters are in place on the installed cleanroom: HEPA filters, lighting, materials of construction, and HVAC units.
- The OQ is performed to verify that the new cleanroom consistently operates within the critical parameters specified. This is typically done per ISO 14644 and typically includes testing for leaks in HEPA filters, total particulate particle count levels, air velocities, differential pressures, and air changes per hour within each room.
- The PQ is a collection of test cases used to verify that a system performs as expected under simulated real-world conditions. PQ involves testing for levels of viable microorganisms in the air as well as on surfaces. In addition to testing for viable organisms, data for total particulates, temperature and humidity are usually collected. The PQ should consist of both static (at rest) and dynamic (in operation) conditions at a minimum, and it is recommended to have multiple data points for each condition to demonstrate consistency.
Qualification and testing
The IQ is the first step of the cleanroom validation. It ensures all critical items that were installed are the correct model and/or type, that they meet the required specifications to make the room function properly, and everything has been installed properly.
Some typical examples that are verified are the types of materials used in the construction of the cleanroom – walls, windows, doors, differential pressure gauges, ceiling panels, flooring, light fixtures, HEPA filters and HVAC systems. All of these components are crucial for the proper functioning of the cleanroom. If any are missing, not the right model, or are not functioning properly, immediate action should be taken to remediate the issue.
For a robust OQ, testing should include particulate concentration, HEPA leak testing, air velocities, differential pressure and airflow visualisation (smoke test).
Determining the number of total particulate samples required for cleanroom validation is not done randomly; it is based on the area of the cleanroom in square meters. Once the area is determined in square meters, table A.1 within ISO 14644-1 is referenced, which dictates the number of locations for each room. The locations of the samples should be based on a grid pattern so they are evenly distributed across the room.

Industry regulatory guidance recommends that in addition to ISO certification testing, the functionality of outlets, interlocks, doors and lights is also verified. After the initial ISO certification is performed during the PQ, it is standard practice to have the room recertified every six months.
Before performing your PQ, perform a risk assessment (RA), which will justify each sample location based on probable contamination sources, product-critical areas, process flows, material flow and personnel traffic.
There are multiple tools suitable for this analysis, however, the failure mode and effects analysis (FMEA) is one of the more commonly used for environmental monitoring analysis.
FMEA starts with building a cause and effect analysis of contamination hazards with an Ishikawa diagram using the Six Sigma 6M approach. Each sample site is then assessed based on potential failure modes of contamination at the particular location, and then a score is applied to three areas – severity, detection and occurrence – to produce a risk priority number (RPN), which then is compared to a matrix with determined ranges for low, medium and high risk.
The PQ sample map is generated from the RA, which will help justify where the sample locations are placed during the PQ, and also for the routine monitoring later on.
Risk-based approach
Industry regulatory agencies such as the US Food and Drug Administration increasingly are requiring the risk-based approach, which should be undertaken by any company planning to validate a cleanroom.
USP chapters <1115> and <1116>, as well as ISO 14644-2, discuss the risk-based approach when developing a sampling plan. Once the sample locations have been determined, the PQ testing can begin.
There can be up to three stages of testing for the PQ. In addition to the at-rest and operational conditions, there also can be the “as built” stage, during which the samples are taken within the cleanroom before any equipment is brought in. In some situations, this is not an option because the equipment is brought into the cleanroom long before the validation work begins due to construction logistics.
Each cleanroom will have a specific ISO classification based on how the area will be used. The three major ISO classifications typically seen throughout biotech, medical device and compounding pharmacies are ISO 5, ISO 7 and ISO 8, with ISO 6 becoming more popular as well.
Each classification has specific criteria that must be met for both the viable and total particulate counts. Of these three classifications, ISO 5 is the strictest, allowing for the lowest amount of viable and total particulates, and ISO 8 is the most lenient, allowing for the highest particulate counts.
Viable acceptance criteria are usually leveraged from the Eudralex vol 4 Annex 1 and the FDA’s Sterile Drug Products Produced by Aseptic Processing – Current Good Manufacturing Practice guidance document.
The latest revisions to USP <1116> have moved away from independent sample location acceptance criteria and toward overall contamination recovery rates. However, for entities that fall under USP <797>, the criteria are still based on a specific number of colony-forming units (CFUs) per plate.
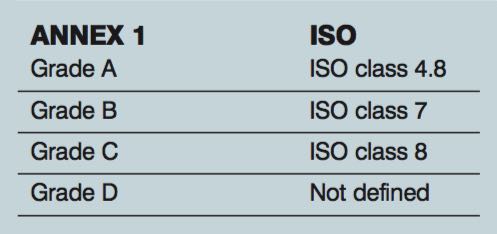
ISO class meets Annex 1
Trying to match up the ISO classifications to Annex 1 can be confusing. Due to the limit of twenty 5.0µm particles for grade A, it does not match up to ISO 5, which has a limit of 29 for the 5.0 µm. In operational conditions, the following is how the ISO matches up with Annex 1.
Contamination levels are controlled by several factors, including, but not limited to:
- HVAC engineering controls such as HEPA filtration
- The cleaning programme
- Gowning
- Aseptic technique training of the personnel
These four elements work in conjunction with each other to maintain your clean environment while environmental monitoring is used to measure the effectiveness of those controls.
The role of monitoring
Environmental monitoring (EM) needs to be performed by trained technicians that have experience with aseptic technique, handling media and sampling equipment, as well as a basic understanding of the concepts of microbiology. This is important to minimise the potential for the technicians to contribute contamination to both the samples and the product during sampling activities.
Once sampling is completed for the day, the media plates should be transported back to the lab for processing. The data from the sampling will then be documented and analysed. The results will be compared against the specifications within your PQ protocol. Representative organisms are typically identified for all growth in your PQ to help establish a working microbial library for your cleanrooms.
Regulatory agencies such as the FDA recommend that a few representative isolates of the microorganisms recovered during the performance qualification are saved and stored. These isolates, or “in-house” microorganisms, can be used as representative facility flora in future studies. The routine environmental monitoring sampling recovery data should be trended for your facility and used to set action and alert limits specific to your operation.
A proper cleanroom validation is critical to ensure products are produced in a controlled, compliant environment. Due to both the complexity and the regulatory importance of a cleanroom validation, it is critical to hire the right team to carefully plan and coordinate each step from IQ to PQ.
Involving experienced partners will make a tremendous difference when it comes to performing the validation properly and in a timely manner. Taking the time on the front end of this process is a great first step to ensuring future success.
This article is featured in the November 2018 issue of Cleanroom Technology. The digital edition is available online.


