
We are all too well aware of the adverse effects of particulate matter contamination within parenteral injectable pharmaceutical products and the consequences for patient safety. Here, particulate matter refers to the small, subvisible particles.
The United States Pharmacopoeia, USP <788> provides two tests for detecting such particulates: light obscuration and microscopic assay. Both are generally accepted for use in testing large volume parenteral (LVP) and small volume parenteral (SVP) for the determination of subvisible particulate matter. Normally, samples are first tested by the light obscuration method. If the sample fails the specified limits, the microscopic assay method can then be used.
Injectable drug products must undergo several discrete processes allowing them to meet or exceed standards developed by the USP that are enforced by the Food and Drug Administration (FDA). Particulate matter in injections is defined as extraneous, mobile, undissolved particles, unintentionally present in the end product.
These contaminants can come from several sources such as the environment, packaging materials, cleanroom personnel and formulation ingredients.
Particulate matter can be extremely harmful when introduced into the bloodstream and can cause adverse reactions in the patient, such as vein irritation, local tissue infarction, anaphylactic shock and even death. Most injectable products are not thermally sterilised, which can present a concerning risk that must be mitigated for patient safety.
USP <788> places limits on the amount of subvisible particles that are allowed in injections. The USP limits on particulate matter, harmonised with the European Pharmacopoeia (EP) and Japanese Pharmacopoeia (JP), are outlined in USP <788> “Particulate Matter in Injections”.
Advances in particle counter liquid sampling technology for light obscuration and sampling software have made the testing process less tedious and more automated.
With the advantage of 21CFR Part 11 (part of the Title 21 in the Code of Federal Regulations that establishes the FDA regulations on electronic records and electronic signatures), sampling software and data integrity with audit trails built-in, particle counter technology is more cGMP compatible. It is essential, however, to understand how this technology meets USP <788> and why injectable drugs manufacturers should use it.
Particle counter liquid sampling: how it works
Light obscuration is the method of choice listed in the pharmacopoeias for the analysis of subvisible particles in parenteral products. The sample is drawn into the system's sensor through a needle or sample tube.
Particles passing a laser beam block a certain amount of light and produce a “shadow” on a light-sensitive detector. The area of this shadow is then converted to the equivalent diameter of the particle.

Liquid Particle Counter used for USP 788 batch sampling
Current methods for light obscuration require a sample volume of 25ml. As to the number of individual units to be tested for LVP and SVP units having a volume of 25ml or more, the USP <788> states that it depends on "statistically sound sampling plans". It further states that: "sampling plans should be based on consideration of product volume, numbers of particles historically found to be present in comparison to limits, particle size distribution of particles present, and variability of particle counts between units." The USP <788> also suggests that the total number of units tested for any given batch may be less than 10 units (for LVP and pooled SVPs) with proper justification.
The liquid particle counter withdraws three samples of not less than 5ml and can calculate the average cumulative counts, average differential counts, average cumulative counts per ml, and average differential counts per ml.
Per the USP <788>, it will automatically omit the data from the first run. The size ranges used for USP <788> testing are ≥10 and ≥25 μm.
Liquid particle counters are capable of sizing and counting particles ranging from 2μm to 200μm.
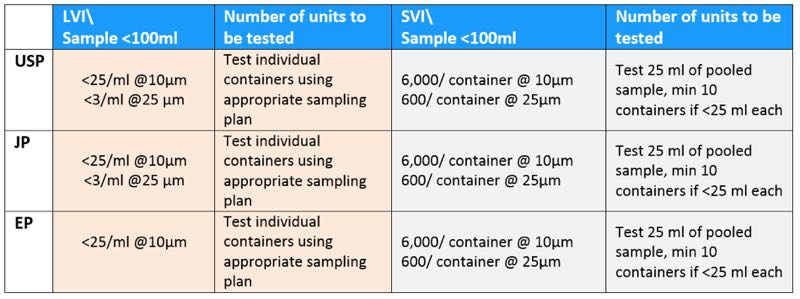
Comparison of USP, JP and EP LVI and SVI sample
USP <788> Validation
Validation of the liquid particle counter is required every six months. The most common validation materials used in the industry are known as Count-Cal particles, which are manufactured by Thermo Scientific.
Count-Cal particles feature the National Institute of Standards and Technology (NIST) traceable diameters, enabling manufacturers of parenteral drugs and injectable solutions to calibrate and document the reproducibility of liquid particle counting instruments.
The Count-Cal particles come with an accuracy of 3000/ml ±10%. The validation testing requires the liquid particle counter undergo several validation tests.
Validation requirements and USP <788>
The Liquid Particle Counter must be calibrated at customer location in-situ and not moved from that location. Calibration and USP <788> validation intervals are on a 6-month cycle.
The basic validation requirements include:
- Sample volume verification
- Volume accuracy test
- Sample flow rate verification
- Sensor calibration
- Sensor resolution test (10µm test particles)
- Count ratio test: blank test using clean, particle-free water
- Count ratio test: 15µm Count-Cal reference particles (3000/ml ±10%)
The verification of the validation is acknowledged when the liquid particle counter passes the count ratio test using 15µm particles. The corresponding counts should fall within 2,700 and 3,300 particles. The validation testing allows for a ±10% tolerance at 3,000 per millilitre. It is a simple but effective validation of the accuracy of the liquid particle counter sensor.
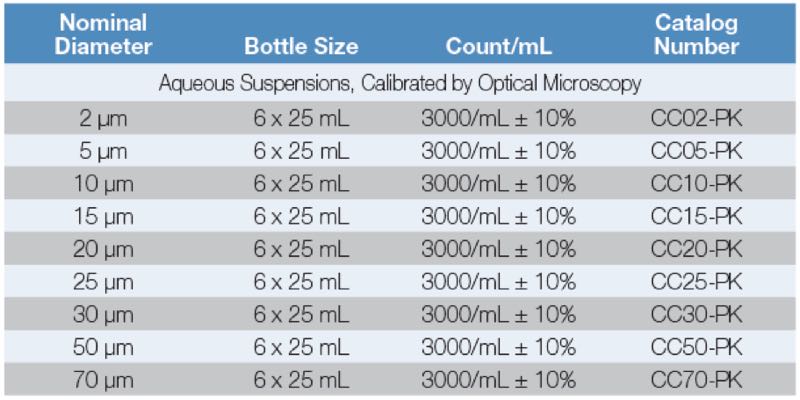
Overview of Count-Cal reference particles used to validate liquid particle counters.
Photo courtesy of Thermo Scientific
In summary, the USP <788> and other pharmacopoeia standards for testing particulate matter are an important line of defence in managing the safety and quality of injectable products.
By building quality testing into the processes, manufacturers can combat the environment in which sterile products are manufactured and keep patient safety a top priority.
References
- USP <788>. Particulate Matter in Injections, United States Pharmacopoeia 37-NF 32, May 1, 2014
- USP <789>. Particulate Matter in Ophthalmic Solutions, United States Pharmacopoeia 37-NF 32, May 1, 2014
- EP <2.9.19>. Particulate Contamination: Sub-Visible Particles, European Pharmacopoeia 5.0, January 2005
- JP <6.07>. Insoluble Particulate Matter Test for Injections, The Japanese Pharmacopoeia 16, March 2011



