
The development of antibody-drug conjugates (ADCs) has provided a new technology platform for the treatment of various cancers and other diseases. In theory, ADCs allow for the targeted delivery of exquisitely potent materials to tumours and other diseased organs, without the concern for off-target toxicity. The development of these new therapies, however, involves handling substances that are among the most potent and toxic to have been handled by any industry.
The ADCs may or may not present the same hazard as the free drug substance and the available data on the different components that make up the conjugated drug product are often limited. This has increased the need for the proper evaluation of the hazards of these new entities to provide the appropriate level of exposure control to individuals handling these materials in the various pharmaceutical work environments and operations required.
During the process of ADC manufacture, several process steps have been identified where occupational exposure can present significant risks. These include synthesis of the payload, conjugation to the antibody, vial filling and lyophilisation.
While a few ADCs have been approved by the FDA1,2 and others are under investigation, the procedures for evaluating and safely handling these compounds have rarely been discussed. Although the adverse effects of occupational exposures to highly toxic pharmaceuticals have been reported in the literature, awareness outside of occupational toxicology and industrial hygiene circles is limited. Standard risk assessment methodologies have historically been used to evaluate and control chemical exposures. However, the extreme toxicity of the materials being handled requires another level of caution in the evaluation of their hazards and control of exposure risks.
Background on ADCs
ADCs may be especially useful for the treatment of aggressive and, frequently, chemotherapy-resistant forms of cancer (e.g. ovarian, prostate and breast cancers, melanoma and leukaemia) due to their ability to deliver very potent and genotoxic drugs directly into cancer cells. This mechanism may improve therapeutic efficacy and decrease off-target systemic toxicity associated with the use of potent payloads, providing an advantage over classic cancer chemotherapy agents.
ADCs typically consist of a humanised antibody (e.g.IgG1-type) conjugated to an active drug (often referred to as the ‘warhead’ or ‘payload’) via a linker (see Figure 1). These three components are further defined in this chapter as:
a) Payload – a highly potent and highly toxic active pharmaceutical molecule, designed (in most cases) to kill the target cell.
b) Linker – a small molecule designed to link the payload to the antibody that will release the payload when it reaches its target (usually a cancer cell)
c) Antibody – selected to bind specifically to an identified disease target, usually a receptor on a cancer cell.
The development of ADC technology has provided the opportunity to revisit the use of certain drugs that were previously deemed too potent or toxic for clinical administration. Among the first ADC payloads developed for cancer therapy were the maytansinoids, or derivatives of maytansine. These compounds are potent, antimitotic agents that act by inhibiting microtubule assembly, which is required for cell replication.3,4 When first discovered, this group of compounds was considered to be one of the most potent in the world, being 100–1000 times more potent than other antimitotic cancer drugs on the market (e.g. vincristine).5 Maytansine was evaluated as a single agent in more than 35 tumour types in more than 800 patients. However, although maytansine was somewhat efficacious against different types of cancers (breast and cervical cancer and lymphoma), the dose-limiting toxicities to the gastrointestinal, blood and nervous systems resulted in discontinuation of all clinical trials.4,6-8
The conjugation of maytansinoids (specifically DM1 and DM4) to antibodies has significantly decreased the systemic toxicity reported in patients. While some adverse effects are still reported, efficacy against tumours is noted below levels causing dose limiting toxicity.
There is a continued effort to create increasingly potent payloads to improve the efficacy of ADCs in cases where antigen binding site numbers or efficiencies may be low. Many new molecules are being investigated as payloads and a few have been approved, including other anti-microtubule agents such as auristatins2, DNA alkylating agents such as pyrrolobenzo-diazepines (PBDs)9 and duocarmycin10, and DNA double-strand breakers such as calicheamicin11. The potency of these molecules varies, but all are highly potent genotoxicants. Therefore, their effects are severe and irreversible, unlike some other highly potent drugs such as peptide hormones, which may be less severe and reversible.
Linker technologies
The linker between the antibody and drug should be designed to enable stability during circulation in the blood, while allowing the rapid and limited release of the active payload inside the tumour cells2. In addition, the conjugate must remain intact during storage in aqueous solution to allow formulations for convenient intravenous (IV) administration. Although research and development in other linkers is on-going, current linkers usually belong to one of two categories: disulfide (cleavable) linkers (see Figures 2 and 3) and thioether (non-cleavable) linkers (see Figure 4)12.
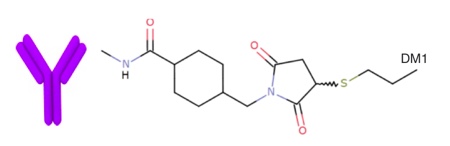
Figure 4 An example of a thioether (non-cleavable) linker
The toxicity of these linkers needs to be evaluated on a case-by-case basis. In preclinical studies of Kadcyla, the thioether linker used in the construction of the ADC did not contribute significantly to toxicity.16
Conjugates with a non-cleavable linker have the advantage of less off-target toxicity in comparison with conjugates with a cleavable linker, as their intracellular metabolism generates amino acid-appended metabolites that are less able to diffuse through cell membranes.17 It is further reported that non-cleavable linkers can result in a single major maytansinoid metabolite that is nearly 50-fold less toxic than maytansine.18 In comparison, disulfide-linked ADCs may result in metabolites that are as little as five-fold less potent than maytansine.
Antibodies
Antibodies, on their own, are generally of relatively low toxicity.19 They are macromolecules usually in the order of 150,000 Daltons, which limits their bioavailability by any route of exposure other than parenteral. The probability of occupational exposure by the typical routes (inhalation, dermal absorption and, to a lesser extent, ingestion) is considered to be relatively low. They are unstable if ingested due to the action of gastrointestinal enzymes and are too large to pass through the skin. Inhalation bioavailabilities of large antibodies such as IgG may be significantly less than 5% of the exposure dose, although that of other antibodies and fragments may be significantly higher.20 The antibody in an ADC is selected to enable highly specific delivery of the payload to an identified disease target. This reduces the risk of undesired effects by ‘carrying’ it until the antibody is bound to the target site where the payload is released. The specificity of delivery enables the payload to be present as a relatively small overall dose (in comparison to traditional chemotherapy agents) further reducing the risk of adverse and off-target effects in the body.
Occupational hazard assessment
Hazard is an inherent property of a material and is not dependent upon factors such as quantity, physical form (e.g. solid, blend or solution, etc.), or potential for exposure. An occupational exposure limit (OEL), which is a quantitative hazard assessment developed for hazardous chemical substances, must be established for the ADC and each of its components. An OEL represents the airborne concentration of a compound which is not likely to damage the health of most workers exposed to those compounds. It is expressed as either an 8-hour Time-Weighted Average (TWA) or 15-minute Short-Term Exposure Limit (STEL). These values must be developed by suitably qualified and experienced toxicologists and are used as a basis to establish appropriate workplace controls.
The traditional approach for determining health-based OELs for substances such as ADCs and their components is to identify a point of departure (No-Observed-Adverse-Effect Level [NOAEL] or Lowest-Observed-Adverse-Effect Level [LOAEL]) from animal or human studies and then to apply appropriate adjustment factors, based on the perceived robustness of the data.21-23 These adjustment factors are applied to account for intraspecies differences, interspecies differences, differences in duration of exposures, dose-response and toxicokinetics issues, and the amount and quality of data available.
Occupational implications and uncertainties
Considering the generic exposure control approach, the risk of injury from exposure to chemicals during their manufacture and handling is a function of:
1. The occupational toxicity (hazard) of the material; an inherent property of the material, and
2. The exposure potential (risk); a variable depending on a number of factors related to the process, the equipment used, the controls in place and the procedures and techniques applied in the handling activity.
When establishing safe working procedures, all potential routes of occupational exposure should be considered including:
- Inhalation by deposition in the lung or by ingestion via the mucociliary escalator;
- Subcutaneous transfer through an open wound, compromised skin barrier, or needle stick;
- Direct absorption through the intact skin;
- Inadvertent contact with mucous membranes (eyes or lips due to contact with contaminated hands/gloves); and
- Ingestion by mechanical transfer from contaminated hands.
While the complete ADC may be considered to have lower toxicity to the patient than the payload, and the bioavailability by occupational routes of exposures is lower, the extreme potency of the payload linked to the antibody still presents a significant hazard. It is important to consider all the above routes of exposure as bioavailabilities may vary from route to route. Although the concept of ADCs is to make the payload unavailable to any cell but the diseased target cell, the ability of that to occur is dependent upon the binding efficiency of the linker technology and the specificity of the antibody. Off-target effects of ADCs represent antibodies that are not 100% specific to target cells. Although these antibodies are usually selected to target cells that over-express certain proteins, there may be other cells that, although expressed to a lesser extent, may still attract these antibodies.
Another area of concern is the presence of unconjugated components as impurities in the final drug formulation. In practice, the relative mass of such impurities may be small compared with the total mass of ADC, and as a result, the weight of hazardous materials will be relatively small even if derived from degradation of the ADC.
Finally, the possibility for ADCs to have local effects in the lung should not be overlooked. Since ADCs are significantly large molecules, it is unlikely that inhalation will result in systemic absorption. However, this does not mean that they won’t have a local effect on respiratory tissue. The potential for off-target or cleaved linkers in pulmonary fluids and tissue exists and therefore, it is often assumed that all the payloads can be released when the ADC is deposited in the lung.
References
1. Hoffmann-La Roche, Ltd. (2013) Product Monograph: KADCYLA® (traztuzumab emtansine). 11-Sep-2013. http://webprod5.hc-sc.gc.ca/dpd-bdpp/index-eng.jsp
2. Seattle Genetics, Inc. (2011) Full prescribing information for ADCETRIS (brentuximab vedotin) for Injection. August 2011. Available at http://www.accessdata.fda.gov/
3. Issell BF and Crooke ST (1978). Cancer Treat Rev 5(4):199-207.
4. Lopus M (2011). Cancer Lett 307(2):113-8.
5. Remillard S, et al (1975). Science 189(4207):1002-5.
6. Tolcher AW, et al (2003). J Clin Oncol 21(2):211-22.
7. Cassady JM, et al (2004). Chem Pharm Bull (Tokyo) 52(1):1-26.
8. Ravry MJ, et al (1985). Am J Clin Oncol 8(2):148-50.
9. Jeffrey SC, et al (2013). Bioconjug Chem 24(7):1256-63.
10. Elgersma RC, et al (2015). Mol Pharm 12(6):1813-35.
11. Damelin, et al (2015). Clin Cancer Res 21(18):4165-73.
12. Erickson HK, et al (2006). Cancer Res 66(8):4426-33.
13. Beck A & Reichert J M (2014) Antibody-drug conjugates: present and future. MAbs 6:15-17.
14. Schumacher, FF et al (2014). Org Biomol Chem 12(37):7261-9.
15. Vedi A & Ziegler DS (2014). Front Oncol 4:82.
16. Poon KA, et al (2013). Toxicol Appl Pharmacol 273(2):298-313.
17. Govindan SV and Goldenberg DM (2010).
ScientificWorldJournal 10:2070-89.
18. Sun X, et al (2011). Bioconjug Chem 22(4):728-35.
19. Hansel TT, et al (2010). Nat Rev Drug Disc 9:325-38.
20. Pfister T, et al (2014). Ann Occup Hyg 58(7):899-911.
21. Galer DM, et al (1992). Regul Toxicol Pharmacol 15(3):291-306.
22. Naumann BD and Weideman P (1995). Hum Ecol Risk Assess 1:590-613.
23. Sargent EV and Kirk GD (1988). Am Ind Hyg Assoc J 49(6):309-13.
In the next issue, Part 2 of this article will cover the establishment of effective controls over exposure.


