
Dr Chris Pickles, CERAM, outlines the main aspects of cleanliness validation for medical devices and compares the different analytical methods that are available to validate cleanliness and cleaning processes.
Residues on the surface of medical devices can cause implant failure and poor device performance. The main source of these residues is materials used in the manufacture of the device, although contamination during the storage, cleaning and handling of the device is also known to occur.
Small amounts of these surface residues can cause deleterious effects in patients, because the residues are in direct contact with body tissues and patients often have compromised immune systems.1 In addition, residues may often alter the surface chemistry and geometry of the device, so even inert residues can be a problem. For example, small amounts of non-toxic cutting fluid on an implant limit the ability of surrounding tissues to attach to the implant.2
To minimise contamination, the US Federal Drug Administration (FDA) stipulates that medical device manufacturers follow specific cleanliness validation procedures.3
First, they must identify all possible residues present on the device and set an acceptable residue limit.4 Then they must use a cleaning regime that reduces residue levels below this limit without leaving significant levels of cleaning agent behind. Finally documentation to verify that residue limits are not exceeded must be submitted to the FDA before the device can go on the market.4
Despite these procedures being in place, some medical devices are failing to meet FDA requirements for cleanliness verification and validation. Since 2001, 173 medical devices have been recalled, some due to contamination issues.5 In just one year of sterility inspections, more than 483 FDA observations related to validation deficiencies.6
Part of the problem for medical device manufacturers and device cleaners is that there are no official residue limits or specified analytical techniques to measure residue levels. The manufacturers have to base their judgments on existing medical devices on the market, which may not be possible if the device has a novel use or uses different materials or manufacturing processes from devices currently on the market.
Another issue is that the surfaces of medical devices are becoming more complex in terms of geometry and chemistry, making cleaning more difficult. Recently there has been a rise in the use of combination devices (devices with both a drug or biologic component and a device component), which creates even more challenges for effective device cleaning. These “combo” devices need to meet quality regulations for both the device and drugs component simultaneously, but the FDA has yet to issue guidance for cleanliness validation for these devices.7
Furthermore, highly aggressive cleaning of the device may produce undesirable surface modifications and inadequate cleaning may leave residues that interact with therapeutic chemicals.7
The advice here is intended for manufacturers of implantables, combo products and of any device that comes into contact with the inside of the body, e.g. theatre instruments, and users of reusable instruments or companies that clean them. Biological contaminants and bioburden analysis are not covered in this article.
Regulatory guidance
None of the regulatory bodies for medical devices stipulates residue limits for medical devices, as absolute cleanliness is unobtainable, nor do they specify the cleanliness validation methods to be used. All of the regulatory bodies stipulate a requirement to identify possible residues, establish a residue limit and to not exceed this limit, and to document and validate cleanliness as part of an ongoing process.8
For example, ISO 14969 requires documented and validated cleaning methods for production facilities, manufacturing equipment, and for the medical devices themselves.8 According to EN ISO 17664, medical device producers are obliged to provide validated and documented methods of reprocessing (cleaning and disinfection) for reusable medical devices.8
The UK’s BS EN46002 standard, a specification for application of ENISO9002, stipulates the following for medical devices: cleanliness of product and validated sterilisation process.8 According to the FDA Guide to Inspections of Validation of Cleaning Processes and The PDA Technical Report No. 29, “Validated cleaning requires a procedure whose effectiveness has been proven by a documented programme providing a high degree of assurance that a specific cleaning procedure, formed appropriately, will consistently clean a particular piece of equipment, device, or area, to a predetermined level of cleanliness – a level objectively substantiated by specific chemical and microbiological tests”.9
Table 1. Types of residues found on the surface of medical devices with examples4,11
| Residues and source of residue | Examples |
| Cleaning agents | |
| Detergents, ionic compounds, acids, alkalis, solvents | Chlorinated cleaners, ethyl and isopropyl alcohol, methyl chloroform (1,1,1-trichloroethane), and trichloroethylene |
| Anionic and non-ionic surfactants | |
| Sterilising agents | Especially Ethylene Oxide (Estrin, 1990) |
| Plastic medical equipment retains formaldehyde residues after low temperature steam and formaldehyde (LTSF) sterilisation (NEWS, 2005) | |
| Packaging | |
| Debris, extractable compounds (plasticizers) | |
| Handling equipment | Dithiocarbamate vulcanization accelerators on latex products (FDA, Technical Guide) |
| Gloves, lotions, skin/oil | |
| Oils/lubricants (hydrocarbon-based) | Alkanes, olefins, additives |
| Lubricants (aqueous-based) | Emulsifiers |
| Inorganics | |
| Grit blast compounds | |
| Loose metal grains | |
| Salt/ionic species | Silica, aluminia, iron |
| Heavy metals | |
| Processing aids | Mould release agents can often be found on plastic devices |
| Polishing compounds (lipid-based) | |
| Dye penetrants | |
| Moulding aids | Silicones, fluorocarbons |
Residues/contamination sources
A problem for manufacturers is the range of possible contaminants/residues for a medical device (see table 1). These residues can react with drug components and other residues to create new, potentially toxic compounds, or can alter the surface geometry of the device by corrosion or by creating layers on the surface of the device (see Fig. 1).10
Contaminants usually fall into one of three categories: water soluble residue, non-water soluble residue and non-soluble debris. Water-soluble residues are usually ionic compounds such as detergents and salts. Non water-soluble residues, such as oils, greases and other hydrocarbons, are soluble in solvents other than water.
Non-soluble debris includes residues such as metals, organic and inorganic solids, and ceramics (Table 2).
Table 2. Chemical classifications of residues4
| Residue Classification | Examples |
| Inorganic | Salts, ceramics and metals |
| Organic (including polymers) | Polar (soluble in water) |
| Apolar (insoluble in water) | |
| Biological | Natural macromolecules |
| Bacterial or viral | |
Residues present as irregular-shaped particles have few attachment points with the surface of the medical device and can be easily removed during cleaning (see Fig. 2).13 Residues that form a single layer on the surface of a device have many more attachment points with the surface of the device and are not so easily removed. However, in the saline environment of the body these residues can be liberated through corrosion reactions and become a potential problem for medical device manufacturers.13 The chemical properties of residues present on the surface of a device help determine the type of analytical method used to validate cleanliness. Residues are sub-divided into different categories (Table 2).
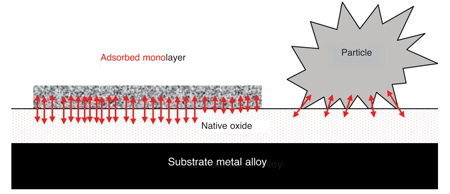
Fig. 2: Comparison of bonding sites for a particle and the same volume of residue adsorbed on the surface of a metallic medical device. Arrows representing bonding sites are not to scale and not representative of the number of bonding sites 22
After identifying the potential residues present, it is the role of the manufacturer to set residue limits and validate that these limits are not exceeded in the manufacturing process. The role of cleaners of reusable devices is to ensure residue limits are not exceeded.
Although there is no official residue limit, there are guidelines for setting residue limits based on the current product qualities: the risk classification of the device, size of the device and the possible residues present.14 For cleanliness validation of a process, the FDA wants to see evidence that the residue limits are logical, practical, achievable and verifiable.6
Table 3: Risk classifications of medical devices used in EU Medical Device Directives6,12,16 (www.MHRA.gov.uk)
| Classification | Risk | Nature of contact and tissue type | Duration of contact | Types of device | Conformity assessment process (including cleanliness validation conducted by) |
| Class I | Low | Surface skin, mucous, membranes | Limited | Examination gloves, tape, blood-pressure cuff, dental dams, endoscopes | Manufacturer/Notified Body |
| Class IIa/IIb | Medium | External communication, Blood, indirect | Prolonged | Dialysis, cardiopulmonary bypass | Notified Body |
| Class III | High | Implant direct | Permanent blood and tissue contact | Stents or grafts, orthopaedic implants | Notified Body |
N.B. The FDA system differs from the EU system in that the devices are classified as I, II, III and IV devices rather than I, IIa, IIb and III
If the device or similar devices are already on the market, it is recommended that manufacturers look at the devices’ history of acceptable performance then use a mean level of residue plus three standard deviations for particulates and other types of residue.15
For new devices, a series of residue spiking biocompatibility studies need to be performed at different levels to determine the failure point of the device.15 At half the failure point, analysis can be performed to demonstrate that the device performance was not affected by the residues present and that toxicity levels were not exceeded.15
If the expected level of residue is known, the device can be spiked at a higher residue level and then evaluated for bio-compatibility and functionality. This higher residue level is the maximum allowable residue limit.
Table 4: Issues concerning cleanliness validation of combo devices7
| Cleaning or assembly issue | Comments |
| Areas proximal to the drug | Cleaning of the device may compromise drug activity |
| Consider materials compatibility | |
| Technical support from cleaning agent supplier is optimal | |
| Corrosion | Avoid chlorine-containing agents with iron-containing alloys |
| Thin films | Films can interfere with drug release or action |
| Particulates | Could interfere with drug delivery |
| Out gassing | Porous materials can absorb and retain cleaning chemicals with unintended slow release |
| Cleaning process design | Balance cleaning action and product modification from wash, rinse and dry |
| Product design | Design for manufacturability including cleaning |
| Be aware of complexities that can trap water or other contaminants | |
| Sterilisation | Heat or radiation may compromise drug portion of the device |
It is also recommended that producers estimate the acceptable daily intake (ADI) for a cleaning fluid or residue, if systemic toxicity based limits are not known. The ADI can be calculated using the LD50 (lethal dose for 50% of the population by compatible route of exposure depending on device) and a conversion factor (usually a value from 100 to 100,000):
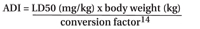
For combination devices, setting residue limits is challenging and a good under-standing of the chemical properties (especially stability) of the biological component of the device is needed; see Table 4 for the issues concerning cleanliness validation for combo devices.
The therapeutic surface chemicals often used to minimise the risk of rejection of implants, such as hydroxyapatite, present an opportunity for interactions between therapeutic surface chemicals and residues.14
Analytical methods
The challenge for the medical device industry in verifying the cleanliness of devices and validating the performance of cleaning processes is to identify one or more measurement techniques that detect the contaminants present on a sample at the required sensitivity.4 The FDA prefers specific methods, although considers any validation method acceptable if it can be justified.18
Once possible contaminants are known and residue limits are set, analysis methods must be chosen that measure the analytes at and below the residue acceptance limits. For this, the Limit of Detection (LOD) (lowest amount of a compound that can be detected) and the Limit of Quantitation (LOQ) (lowest amount of compound that can be quantified) of the analytical tool, need to be established. The residue acceptance limit should be well above the LOQ, so that the residue can be accurately quantified.19
To satisfy the FDA’s cleanliness validation criteria, the FDA needs to be assured that the validation process is repeatable, the process does not adversely affect the product or the packaging, and the process meets the sterility assurance limit (SAL) in the worst-case scenario. (The SAL is the probability that an implant will remain non-sterile following sterilisation – one in a million.)4,20 The FDA recommends that the following criteria are considered before implementing an analytical procedure for a cleaning validation application.4
Sensitivity – the method is appropriate for the residue limits in terms of sensitivity and LOD of the device.
Practicality – the method is practical and rapid and, if possible, uses established pre-existing techniques and equipment.
Validation Scheme – the method is readily validated in accordance with regulatory requirements for instrumentation.
Successful Recovery Study – the method should include compound recovery studies that challenge the sampling and testing methods.4
The analytical methods can be subdivided into two categories: direct and indirect. Direct methods detect the residue directly on the surface of the device; indirect methods require residues to be extracted prior to analysis. For indirect methods, residues are extracted by washing the device with water, aqueous solution or an organic solvent and then collecting the rinse water, or by direct surface sampling with a swab. Exceptions to this are volatile organics or absorbed gases, e.g. ethylene oxide, which are extracted via thermal evaporation using a headspace sampling technique.
At the very minimum, the method requires two different analysts, instruments, columns (if chromatography is being used), days for experimentation, and the use of prepared samples and standards.19
The different cleanliness validation methods are divided into three main categories: direct surface analysis, residue analysis and gravimetric analysis. Both gravimetric analysis and residue analysis are indirect methods; of course, direct surface analysis is a direct method.
The choice of method depends not only on the anticipated residues present and residue acceptance limits, but also includes a consideration of the pros and cons of the methods, along with cost considerations. Usually more than one method is used to validate the cleanliness of the device to ensure that the entire range of possible contaminants is detected.
Of all the methods, surface analysis techniques are the only ones that can identify the location of the contaminant on the device and detect extremely low levels of residue. So, the surface analysis techniques can potentially be used to identify the manufacturing stage or process in which contamination has occurred. Two surface analysis techniques that are often used to validate cleanliness are ToF-SIMS and XPS because they analyse the outermost surface layers and therefore directly measure residual contamination.22
All of the direct surface analysis techniques examine the top few layers of the device’s surface by irradiating the sample with x-rays or ion beams and analysing the emitted electrons or secondary ions. In contrast, the indirect residue analysis methods use a variety of techniques to detect residues present on the surface of the device. Descriptions of how the different methods work are given in the full white paper.*
Gravimetric analysis is an indirect analysis technique. The device is placed in an extraction media and the resulting eluant is passed through a pre-weighted membrane filter. The filter is then dried and re-weighed. It is assumed that any difference in mass is due to particles present on the surface of the medical device and these particles are of the same density and size as the original tests used to set acceptable mass levels for the residues.
Benefits of validation
There are numerous benefits to the manufacturer for conducting a robust cleanliness validation procedure:
- Minimise time and costs of protocol submission
- Maximise product revenues – product can be sold while the FDA reviews files
- Optimise the producer’s competitive situation – they can think about how their submission type and structure will affect their competitors and suppliers
- Reduce or eliminate product loss, recall and rework caused by contamination failures.1, 21
For manufacturers and cleaners of medical devices, CERAM offers a range of independently verified cleanliness analysis techniques particularly focusing on surface analysis measurements. The company has developed the ‘Validata’ cleanliness index (CI) which expresses surface cleanliness as a single figure ranging from 0 to 100% derived from a complex combinatorial algorithm.18 This index enables a simple comparison between samples and permits the monitoring of process trends; the index also enables the easy communication of surface cleanliness validation findings.
Of the various analytical methods available to measure device cleanliness, and thereby truly validate the cleaning process, only surface analysis can inform the level of adsorbed contaminants.
References
1. D. Beal. Medical Device and Diagnostic Industry, 2006, Aug 8-12
2. M. Jackson and W. Ahmed. Surface Engineered Surgical Tools and Medical Devices. 2007 Springer
3. FDA 2003: http://www.fda.gov/MedicalDevices/DeviceRegulationandGuidance/GuidanceDocuments/ucm089726.htm#_Toc33587914
4. R. Luginbuehl, B. Gasser, and V. Frauchiger. Journal of ASTM International. 2006, 3 (5), 1-11
5. Medical Device Recalls (2009): http://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfRES/RESSimpleSearch.cfm?start_search=131&Search_Term=contamination&PAGENUM=10&sort=RECALL_INITIATION_DT%20DESC
6. A. Booth, Sterilization of Medical Devices. 1999 Interpharm Press
7. E. Kanegsberg, B. Kanesberg, and J. Philips, Controlled Environments Magazine, 2008. Mar, 1-6
8. A. Daniel, E. Kimmelman, and K. Trautman, The FDA and Worldwide Quality System Requirements Guidebook for Medical Devices. 2008 Quality Press
9. R. Brunkow et al, Cleaning and Cleaning Validation: A Biotechnology Perspective. 1996 Interpharm/CR, Chapter 2: Cleaning Mechanisms and Strategy, 41-51
10. B. Kanegsberg and E. Kanegsberg, Controlled Environments Magazine, 2006, Sep, 1-5
11. S. Spiegelberg. Current Activities in Cleanliness of Biomedical Devices at ASTM 2003: http://www. campoly.com/documents/appnotes/astmshs.pdf
12. R. Riechl, Int. Journal of Sterile Supply. 2003. 11 24-25
13. L. Hazell and J. Canry. Limitations on the Production and Maintenance of Clean Surfaces. 2009
14. D. LeBlanc. Journal of ASTM International. 2006 Mar, 1-4
15. J. Broad and B. Kanegsberg, Controlled Environments Magazine, 2007 Sep, 10-20
16. D. Albert. Medical Device Technology. 2004, 1-4
17. M. Jackson and W. Ahmed W. Surface Engineered Surgical Tools and Medical Devices. 2007 Springer
18. M. McLaughlin and A. Zisman. The Aqueous Cleaning Handbook – A Guide to Critical-cleaning Procedures, Techniques, and Validation: 2002 http://www.alconox.com/downloads/pdf/cleaning_handbook_3rd_edition.pdf
19. H. Kaiser and M. Minowitz. Journal of Validation Technology, 2001 May Vol 7, (3)
20. E. Arscott et al. Medical Device and Diagnostic Industry, 1996. 1-12
21. C. Pickles, Pharma. Tech. Eur., 2008 Nov, 49-56
22. CERAM private communication.
*The full version of this White Paper can be found on the CERAM website at http://www.ceram.com/ newsroom/news-releases/cerams-new-white-paper-analyzes-cleanliness-validation/


