
Those who work in cleanrooms know it well: avoiding cross-contamination between rooms with incompatible cleanroom classifications is essential to preserve the desired ISO classification and maintain the integrity of products and processes. One way to achieve this is by minimising material movement.
Non-viable particles and microorganisms can cause contamination in a cleanroom due to this entry and exit of materials within the controlled environments. With this in mind, it is necessary to design and correctly install material transfer equipment.
A pass box, which is also called a transfer hatch or SAS pass (sterile access system), works as a barrier between areas with different levels of cleanliness when materials do have to be moved. The equipment is used to transfer material from an area of lower cleanliness to an area of higher cleanliness, and vice versa.
All cleanroom pass boxes include a mechanical and/or electrical interlock system designed to minimise the risk of cross-contamination. This design ensures that only one door can be opened at a time, not both or more, thus minimising the amount of “dirty” air that can enter the cleanroom.
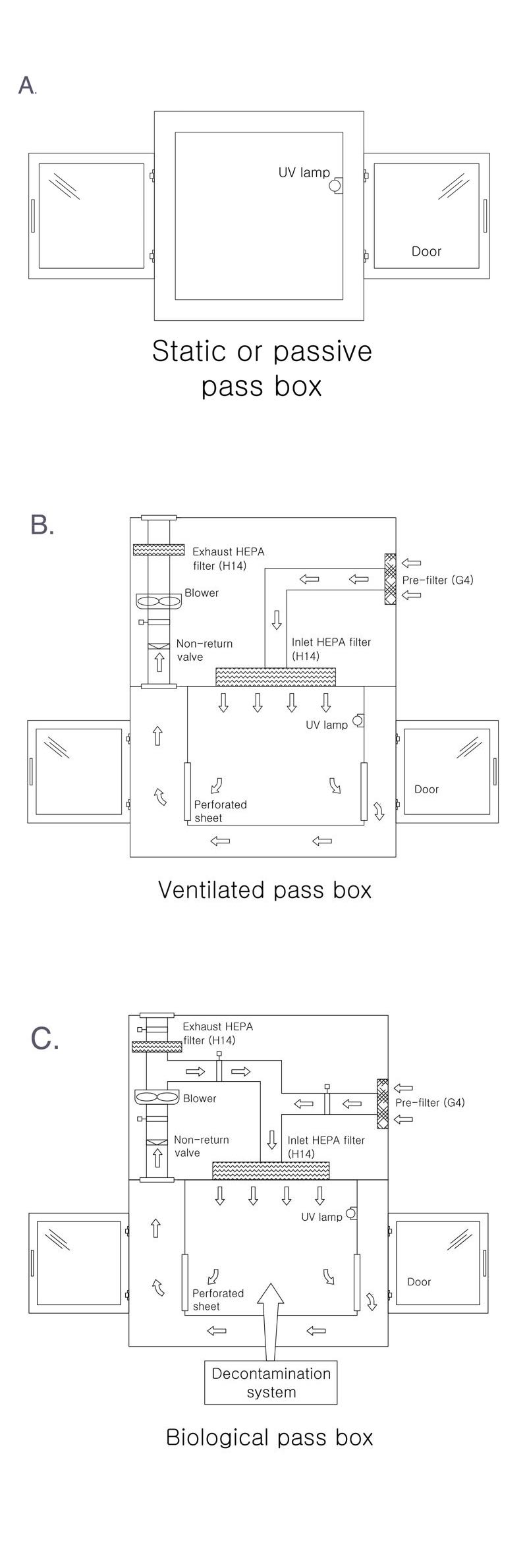
Usually, pass boxes have a UV light lamp in them to remove the contamination that may enter during the transfer of material.






