The ICH Harmonised Tripartite Guideline Validation of Analytical Procedures: Text and Methodology Q2(R1) 2005 and its predecessors, 1994 and 1996, have been the global regulatory standard for almost a quarter of a century. Indeed, their origins are older than that as it was broadly based on USP General Chapter <1225>, which predates it.
It was written originally to provide guidance on how the ‘new’ chromatographic procedures, particularly HPLC, should be demonstrated as ‘fit for intended’ use as well as the traditional methodologies under a unified approach as shown, for example, in Figure 1.
The regulatory and industrial expectation of seeing ‘validation’ as an event with associated documentation was born with a strict set of analytical performance parameters, which had to be determined and documented.
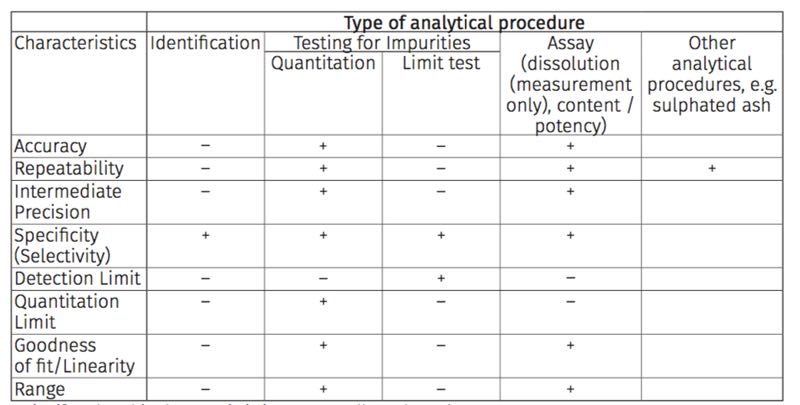
Figure 1 Traditional Analytical Performance Characteristics required by analytical procedure type
- signifies that this characteristic is not normally evaluated
+ signifies that this characteristic is normally evaluated
Regulatory Environment
Much has changed in the intervening years. In particular, the concept of process validation has changed from the original FDA Process Validation Guidelines of 1987 definition of;
“Process validation is establishing documented evidence which provides a high degree of assurance that a specific process will consistently produce a product meeting its pre-determined specifications and quality characteristics”
to the revised FDA Process Validation Guidelines of 2011 definition of;
“Process validation is the collection and evaluation of data, from the process design stage throughout production, which establishes scientific evidence that a process is capable of consistently delivering quality products”
Hence it is seen that validation is no longer considered an activity but rather a sequence of events over a lifecycle. Similar evolution has occurred in EU GMP Annex 15 on Validation.
Since 2005, there have been significant changes in both regulatory expectations within ICH, the GMPs and the Pharmacopoeias as well as analytical practice within industry.
For example, in ICH, we have seen not only an expansion in the membership from the original three to 16 ICH Members and 27 Observers in 2018 but also new guidelines;
- ICH Q8(R2), Pharmaceutical Development, August 2009
- ICH Q9, Quality Risk Management, November 2005
- ICH Q10, Pharmaceutical Quality System, June 2008
- ICH Q12: Technical and Regulatory Considerations for Pharmaceutical Product Lifecycle Management, Step 1 Core guideline, June 2017
FDA Analytical Procedures and Methods Validation for Drugs and Biologics Guidance for Industry; July 2015 has been updated to include a reference to lifecycle methodology.
USP has been active in providing updated general chapters;
- USP 40 (2018), General Chapter <1224>, Transfer of analytical procedures
- USP 40, (2018), General Chapter <1225>, Validation of analytical procedures
- USP 40, (2018), General Chapter <1226>, Verification of analytical procedures
And a new initiative in 2013
- Lifecycle Management of Analytical Procedures: Method Development, Procedure Performance Qualification, and Procedure Performance Verification
- Proposed New USP General Chapter: The Analytical Procedure Lifecycle <1220> in 2017
Resulting in a
In 2018, ICH has announced the revision of Analytical Procedure Development and Revision of Q2(R1) Analytical Validation (Q2(R2)/Q14) within their next review cycle.
Analytical control practices in industry
When ICH Q2 was first developed, the majority of analytical procedures were carried out in the laboratory. Since that time, many identifications take place in the warehouse or production as well as in in-process controls.
Many of these measurements are based on spectroscopic methodologies such as NIR (Near Infrared) or Raman techniques. In addition, new sensor technologies are also being employed as part of PAT (Process Analytical Technology). It has long been recognised that the ICH Q2(R1) model is not adequate to cope with ‘fitness for purpose’ determination of such procedures.
A redefinition of the term validation is required because “validation” does not only address the determination of each validation parameter at a single point in time with respect to its variability, but should also be extended to a continuous process
Hence it can be seen that, the current ICH validation guideline does not adequately cover;
- Validation parameters for other analytical techniques, e.g. NIR or Raman, procedures for the determination of elemental impurities, e.g. ICP-OES, ICP-MS
- Acknowledgement of the fact that the reportable result has an uncertainty budget associated with the precision and accuracy of the method
- Adequate terminology e.g. the term “specificity” is not appropriate as the text describes more “selectivity”, i.e. identification of the analyte in the presence of matrix components
- Guidance on how to set appropriate acceptance criteria for the validation parameters
- Information with respect to requirements for a stability-indicating procedure
- An adequate description of non linear fitting models
- Two fundamentally different aspects which are confused
- The “ability to obtain test results which are directly proportional to the concentration (amount) of analyte in the sample” refers to the reportable result of the analytical procedure but is already included in accuracy.
- The relationship of “signals as a function of analyte concentration” refers to the analytical response function, i.e. the calibration model, which is often the linear one described in the guideline but may assume any other mathematical model.
- Statistical basis for the determination of the sample size for each validation parameter
- The examples given for the calculation of detection and quantification limits are based on the slope of a linear calibration model and thus cannot be used when the calibration model is non-linear
- A redefinition of the term validation is required because “validation” does not only address the determination of each validation parameter at a single point in time with respect to its variability, but should also be extended to a continuous process in order to control the analytical procedure under life-cycle conditions.
Data governance and data integrity
In recent years, there have been major regulatory concerns regarding data governance and data integrity as regulatory inspections have uncovered many instances of data falsification and fraud. However, the large majority of data integrity breaches are more attributable to poor or bad practices and lack of adequate analytical controls than deliberate falsification.
It is important to understand that data governance and data integrity are concerned with more than just the generation and security of correct numbers. Data Governance is concerned with the totality of arrangements under a Pharmaceutical Quality System (PQS), to ensure that data, irrespective of the format in which they are generated, are recorded, processed, retained and used to ensure complete, consistent and accurate records throughout the data lifecycle, and product lifecycle.
Clearly, therefore, there is also a need to control and monitor the means of generating analytical data, both technically and procedurally. The analytical procedures themselves require a lifecycle management approach to ensure that they are initially ‘fit for intended purpose’ and continue to perform within their design capabilities throughout their working lives.
ECA Analytical Quality Control Group (AQCG)
Established in 2010 as part of the ECA Foundation, the Group objectives are;
- Provision of a European wide networking platform for Analytical Chemist and Scientists working in and managing a Quality Control environment.
- Promotion of active discussion of the latest regulatory requirements for Quality Control within the European Union and US.
- Identification and addressing of technical issues and challenges within the Quality Control environment including training needs.
- Active support for a harmonised approach to common problems and issues by the generation of discussion/position papers and generic procedures via expert working groups.
- Facilitation of effective and efficient communication between industry, competent authorities and the pharmacopoeias.
Much effort has already been made in meeting objective 4 in particular by 2017 which has resulted in the generation of three guideline documents proposing harmonised approaches available to all ECA members;
- OOS Guideline, v2.1, August 2013
- OOE & OOT Guideline; v1.1 November 2016
- Data Governance and Data Integrity Guideline; v2.0 January 2017
However by early 2017, it had become apparent that there was a need to produce a detailed practical guideline attempting to consolidate and harmonise the life cycle approaches for analytical procedures.
New ECA AQCG Guideline on Analytical Procedure Lifecycle Management (APLM)
The purpose of this ECA AQCG guideline is to provide;
- An overview and framework for a lifecycle approach to the qualification of analytical procedures as part of a lifecycle approach which is consistent with the principles of the FDA process validation guideline and regulatory expectations. See Figure 2
- Descriptions and recommendations regarding the staged activities in the lifecycle
- Input for the forthcoming revision of ICH Q2(R1), the new Q14 and support of the on-going development of USP General Chapter <1220> on Analytical Procedure Lifecycle
- Guidance on the application of the lifecycle approach for existing methods and procedures
- A focus for debate regarding the practical implementation of APLM within the industry
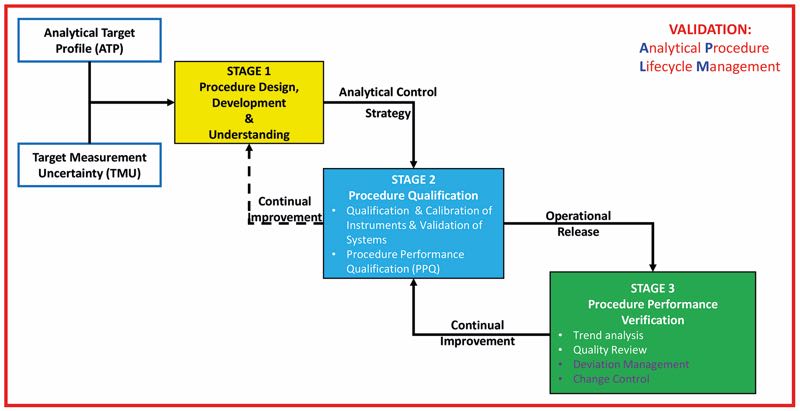
Figure 2: An overview and framework for a lifecycle approach to the qualification of analytical procedures a lifecycle approach
This guideline is intended to apply to chemical methods of analysis for both univariate methods such as HPLC, UV spectrophotometric assays for example and multivariate methods for identification of materials using FTIR, FTNIR and Raman spectroscopies for example.
Univariate is a term commonly used in statistics to describe a type of data which consists of observations on only a single characteristic or attribute whereas multivariate encompasses the simultaneous observation and analysis of more than one outcome variable.
These principles also apply to biopharmaceutical assay methods. Biological and microbiological methods are not explicitly covered by this guideline.
The key areas covered in the guideline include;
- Principles of Analytical Procedure Lifecycle Management (APLM)
- Prerequisites for the APLM
- Guidance recommendations for the 3 stages analytical procedure of the APLM
- Stage 1: Procedure design, development and gaining process understanding
- Stage 2: Procedure Performance Qualification
- Stage 3: Procedure Performance Verification
- Six technical appendices
A full day workshop session and launch of this new 80 page guideline will take place during the PharmaLab Congress 2018 in Dusseldorf. For more detailed information please visit www.pharmalab-congress.com.