
Biotechnology is a future-orientated industry and its presence in the marketplace is increasing. There might have been setbacks caused by stock market crises and temporary cautious investment volume, but the marginal increase on R&D to €1.1 billion in Germany, for example, is a testament of the upward trend.
The increased importance of biopharmaceuticals can also be recognised by the proportion of biopharmaceutical products among the top sellers: two-thirds of the world's best-selling medicinal products are biopharmaceuticals, in particular, new therapies. Tissue preparations and gene therapeutics have become significantly more important in the recent past and have made it necessary to issue new or revised regulations.
The basis for GMP requirements, also for companies in pharmaceutical biotechnology, is Union DIR (EU) 2017/1572/ by the European Union. Several other directives define the specifications for individual product types. Among them are DIR 2001/83/EC (medicinal products for humans), DIR 2001/82/EC (veterinary medicinal products), DIR 2002/98/EC (blood and blood products) or DIR 2004/23/EC (tissue and cell products) and, more recently, the regulation for novel therapies (ATMP) 1394/2007.
Moreover, guidance regarding requirements for medicinal products is also provided by directives and guidelines from professional bodies. These include the Notes for Guidance/Guidances of the European Medicines Agency, but also the EU GMP Guide with Parts I-III and the Appendices, describing the requirements for specific product groups or manufacturing areas in more detail. These documents serve to interpret the legal requirements and usually represent the “state of science and technology” (Fig.1).
On an international level, the following guidelines are also important for biotechnology:
- FDA Biotechnology Inspection Guide (1991)
- FDA Guide for Industry: Bioanalytical Method Validation (2001 and draft revision 2013)
- ICH Guidelines, e.g. Series Q5 A-D, Q6 B, Q11
- EU GMP Guide, Part II (GMP for APIs, Chapter 18)
- PIC/S documents, e.g. PI 024-1 'Aide mémoire Inspection of Biotechnology Manufacturers' (Dec. 2005)
- ISPE Guides, e.g. Vol. 6 ‘Biopharmaceutical Manufacturing Facilities’ (2013) and Process Development and Manufacturing, (2013)
- PDA Technical Reports, e.g. B. No. 57 Analytical Method Validation and Transfer for Biotechnology Product Single Use Systems (2014), no. 74 Reprocessing of Biopharmaceuticals (2016) and no. 66 Application of Single- Use Systems in Pharmaceutical Manufacturing
European framework
Throughout the EU, the EU GMP Guide with its 19 annexes are valid as the detailed interpretation of the principles of good manufacturing practice defined in the Directives (e.g. Directive 2003/94/EC, Directive 2001/83/EC) and following national regulations.
The European Union’s pharmaceutical legislation is built on four pillars:
- The classical medicinal products for humans
- Veterinary medicinal products l Blood and blood products
- Human tissues and cells
For each area, there are directives providing the provisions for national legislators and will be interpreted in documents as mentioned in Fig.1.
In its two parts, the EU GMP Guide defines the general requirements for good manufacturing practices for active substances and medicinal products, but not differentiating between chemical, synthetic and biological products. There is no focus on biotechnological aspects, except for Chapter 18 in Part 2.
In the centre of Part 1 are authorised medicinal products and non-clinical test material. Part 2 of the Guide addresses the essential requirements for APIs for use as starting material. In Chapter 18 of Part 2 there are special references for material produced by cell culture/fermentation.
Annex 2 of the EU GMP Guide (2012) is dedicated to the requirements of biological drugs and active ingredients. It provides the guideline for regulatory inspections, as it also gives inspectors detailed guidance and interpretation of the requirements.
An important change is evident in the title of Annex 2, which has been extended from “Manufacture of Biological Medicinal Products for Human Use” to “Manufacture of Biological Active Substances and Medicinal Products for Human Use”. This means that the Annex now also applies to manufacturers of biopharmaceutical APIs.
The concept of quality risk management (QRM) – the detailed risk analysis for individual production steps – shows throughout the entire document. It reads: “... quality risk management (QRM) principles are particularly important for this class of materials and should be used to develop the control strategy across all stages of manufacture.”
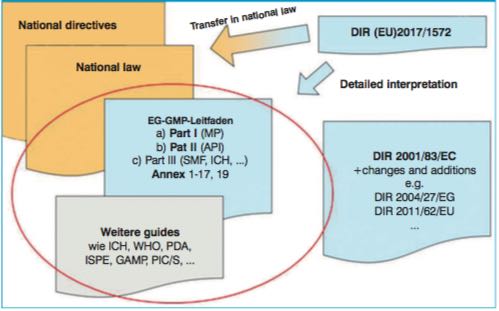
Figure 1: Overview and classification of the various regulatory documents. Example documents for the current state of science and technology are circled
Unique challenges
To meet the changing requirements and the diversity of today's biopharmaceutical products, Annex 2 includes two big sections – among others. Part A General guidelines and Part B Specific guidelines for selected product types.
Part A contains the general information for the manufacture of APIs and products as well as information in regards to personnel, cell banks and final production. Part B also provides information on selected types of biopharmaceutical agents and finished products.
The general structure has remained the same in comparison to the previous version of Annex 2. However, when looking at the individual chapters and their sub items one can notice a much larger scope of requirements. Some examples of this would be:
- The chapter on Personnel focuses on close monitoring of health. There is also a requirement that after working in areas where genetically engineered organisms have been used, staff should not be working in areas of other products on the same day without special precautions against cross-contamination.
- The chapter on rooms and equipment provides information on the scope of environmental monitoring, on dedicated multipurpose production, and on the qualification of rooms and equipment. It is emphasised that contamination prevention is more important than monitoring and removing contaminants.
Part B of the new Annex 2 deals with specific requirements for certain biopharmaceuticals, depending on their origin or type of product. This section includes the following groups:
- Products of animal origin l Allergenic products
- Animal immune sera
- Vaccines
- Recombinant products
- Monoclonal antibodies
- Transgenic products of animal origin
- Transgenic products of plant origin
In the coming necessary revision, the following groups will be removed from the Annex 2 as they will be regulated by the new ATMP Guidelines:
- Gene therapy products
- Somatic and xenogenic cell products and tissue products
Quality standards for marketing authorisations
The “New Guidelines on Good Manufacturing Practice specific to Advanced Therapy Medicinal Products” will become effective in May 2018. In the new document, the authors state that GMP is required for all medicinal products with a marketing authorisation. Moreover, for products manufactured under hospital exemption an equivalent quality standard is required. They further emphasise that the development of such specified guidelines for ATMP are mandated by Article 5 of Regulation (EC) No 1394/20072 and by Article 63 (1) of Regulation (EU) No 536/2014 for IMPD.
The new guideline includes requirements for ATMP with a marketing authorisation as well as for advanced therapy medicinal products that are being tested or used as reference in a clinical trial (i.e. advanced therapy investigational medicinal products).
The document should be the official paper for the definition of the GMP requirements for ATMP, so in the scope it states: “These Guidelines do not apply to medicinal products other than ATMPs. In turn, the detailed guidelines referred to in the second paragraph of Article 47 of Directive 2001/83/EC4 and Article 63 (1) of Regulation (EU) No 536/2014 do not apply to ATMPs, unless specific reference thereto is made in these.”
The table of content shows that the status as a stand-alone guideline has made it necessary to include all important fields with relation to ATMPs, which are normally covered for other medicinal products in the existing GMP guideline:
- Risk based approach
- Personnel
- Premises
- Equipment
- Documentation
- Starting/Raw Materials
- Seed lot /call bank systems
- Production
- Qualification and Validation
- QP and batch release
- Quality Control
- Quality defects and recalls
- Reconstitution
- Automated production
Topics such as hygiene aspects as well as aseptic processing requirements and finally batch release issues are all covered in several bullet points of these chapters.
For manufacturers of ATMP, it will now be the challenge to check whether the current established processes will be in accordance with the new guideline or whether some changes or additional activities will be necessary.
Further information on current ATMP topics and events is available online at www.atmp-group.org.
Editor's note: This article appeared in the April issue of Cleanroom Technology. The digital edition is available online.






