Organisations planning for allogeneic cell therapy donor collections typically expect Good Manufacturing Practice (GMP) requirements will mean collections must occur in a cleanroom. It sounds intuitive, but it’s wrong.
Although cellular starting material collections is critical for manufacturing, the facility standards required for open processing steps do not apply equally to donor aphaeresis. Real-world evidence, the regulatory framework, and decades of practice in transfusion medicine all point in the same direction: cellular starting material can be collected safely, reproducibly, and compliantly in well-controlled environments without cleanroom classification.
The misconception is understandable, as aphaeresis certainly should be performed in a space with intentional design, defined controls, validated cleaning, disciplined behaviour, and consistent execution. A cleanroom fits this category but is not, by itself, the only way to achieve these requirements.
The process’ safety profile is determined by how it is designed, maintained, and used. When those same physical and protocol‑driven controls are deliberately applied to donor collection, a controlled, non‑classified donor room meets the same regulatory intent without cleanroom infrastructure.
It is important to examine the operational and regulatory drivers that determine contamination risk, and understand how leading collection centres achieve compliant, high-quality starting material using a control framework that has supported the blood banking industry for over half a century.
What collection standards actually require
In cell therapy manufacturing, cleanrooms are essential when activities involve sustained exposure of cells or reagents to the environment—including steps such as viral vector handling, cell expansion or culture, washing, formulation, or cryopreservation, where product is open, and environmental controls are required to protect quality and prevent contamination.
Donor collection is fundamentally different. In the US, the regulatory standard is based on 21 CFR Part 1271, which governs FDA’s Good Tissue Practice (GTP), as well as long-standing industry standards as defined by the Foundation of Accreditation of Cellular Therapy (FACT), and through the Joint Accreditation Committee (JACIE), which harmonises European and US requirements.
When read closely, both FACT-JACIE Standards for Collection Facilities and GTP regulations describe expectations for controlled environments and documented risk management, but neither prescribes cleanroom design for donor collection activities.
Under the more specific FACT-JACIE standards, collection is required to occur in a controlled and suitable environment, supported by documented risk assessments, procedures to prevent contamination and mix-ups, and environmental monitoring based on risk-assessment. The standards emphasise intentional facility design, segregation where necessary, and executing of defined processes, not air classification.
Cleanrooms are not specified as a categorical requirement for donor collection; rather, collection programmes are expected to demonstrate that their environment is appropriate for the activities performed and that risks are understood, mitigated, and justified. FDA has the same expectations under 21 CFR Part 1271. Nowhere in Part 1271 does FDA prescribe ISO classification, HEPA filtration, or cleanroom construction requirements for donor collection. Environmental controls are expected to be aligned with the risk profile of the process, not imported wholesale from manufacturing standards.
A recurring source of misunderstanding is regulatory vocabulary drift, in which GTP‑regulated collection is implicitly treated as a scaled‑down form of cGMP manufacturing. This is not how the regulations are structured. Manufacturing requirements under 21 CFR Parts 210 and 211 are triggered by open product exposure and manipulation that alter or directly affect the product.
Donor collection, when performed using closed or functionally closed systems, is intentionally regulated under a different framework. The absence of a cleanroom requirement for collection is not an omission; it reflects a deliberate, risk‑based distinction embedded in both FDA regulations and accreditation standards.
Collection is not manufacturing
FDA regulations recognise aphaeresis-based collection as a procurement activity, not a manufacturing one. Under 21 CFR 1271 Subpart D, donor collection responsibilities include:
- Verifying donor eligibility
- Using sterile, single use, closed system collection kits
- Following defined procedures to prevent contamination and mix-ups
None of these requirements trigger cGMP manufacturing obligations in a classified space. In fact, manufacturing under §1271.3(d) is defined specifically as manipulation that alters biological characteristics—something donor collection simply does not do.
Closed, functionally closed, and open workflows: Why that matters
Table 1 explains the difference between these three terms.
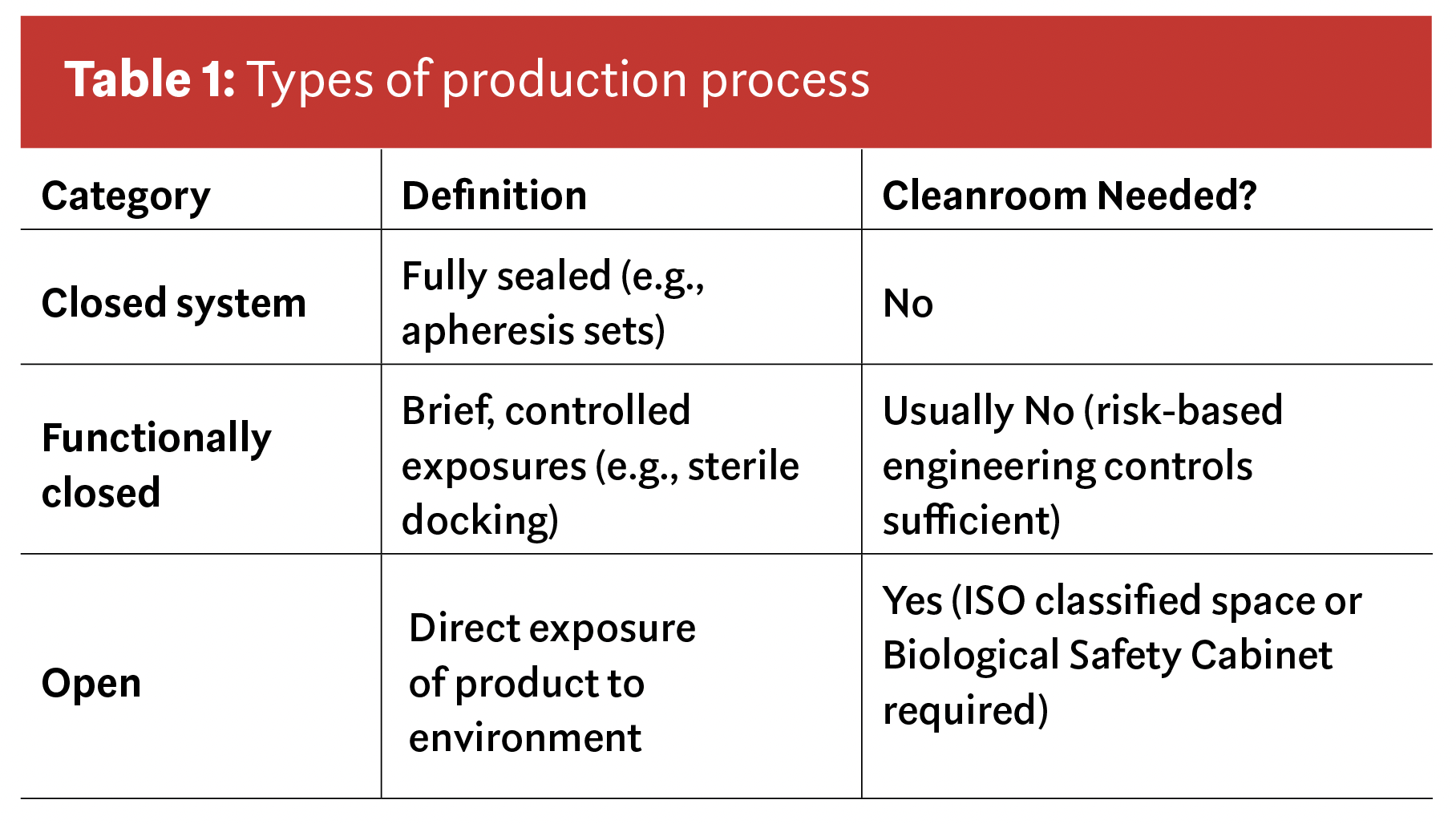
Nearly all modern cellular collections fall into the first two categories; closed and functionally closed. That alone changes the environmental control discussion dramatically. It also clarifies why facility standards designed for open manufacturing steps do not automatically apply to donor collection.
Behaviour and process, not walls
FDA’s GTP framework is unapologetically risk-based. It does not prescribe air classifications, HEPA systems, or ISO zoning for collection. Instead, it focuses on how work is done.
Operator behaviour remains the dominant contamination risk, even in highly engineered environments. The focus is on competency‑based aseptic technique, hand hygiene, proper skin preparation, disciplined line management, and adherence to defined procedures rather than improvisation. A well-designed donor room with highly trained staff outperforms a poorly run cleanroom.
Facility and equipment controls are intended to be fit for purpose, not over‑engineered by default. The collection environment must be appropriate for the work being performed, cleaning and disinfection practices validated, aphaeresis equipment properly maintained and calibrated, and environmental conditions controlled in a way that reduces contamination risk. These requirements are functional, not architectural. What matters is that the space supports disciplined work optimised for successful procurement with minimal risk for contamination, not that it resembles a manufacturing suite.
Materials management and documentation provide the quality backbone that makes controlled collection environments reliable. Donor collection should rely on sterile, single‑use kits, contamination‑mitigating engineering controls such as initial specimen diversion, full traceability of consumables, and supplier qualification. Robust chain‑of‑identity and chain‑of‑custody practices, deviation management, and CAPA under §1271.270 ensure that risks are detected, addressed, and prevented from recurring. Contamination control in this context is achieved through systems and execution, not through air classification. Cleanrooms are simply one possible way to enforce behaviors and processes that GTP expects.
Environment by risk
A controlled donor room is intentionally designed for blood collection, with restricted access, trained staff, validated cleaning practices, and standardised workflows centred on closed or functionally closed collection systems.
Unlike surgery, which requires a sterile environment due to extensive tissue exposure, donor collection involves only a brief skin breach at venipuncture. The limited risk is mitigated through:
- Aseptic technique
- Proper site selection and skin prep
- Sterile, single-use collection devices
- Engineering controls, such as the sample diversion pouch, which captures the initial blood most likely to contain residual skin flora
In donation, the closed collection system is the primary contamination barrier. It isolates the blood product from the environment, allowing environmental controls to be appropriately scaled to the actual risk rather than treating the entire room as a cleanroom.
The donor room’s purpose is not to ensure sterility of the entire space but to support consistent, disciplined execution of these risk-based controls.
When designed and operated intentionally, a controlled, nonclassified donor room can meet the same regulatory intent often attributed to cleanrooms, without unnecessary classification.
Designing for GMP product quality at scale
Cellular starting material collection is ultimately a question of how best to protect product quality at scale. For donor collection, protection is achieved not through environmental sterility, but through closed systems, disciplined execution, and layered controls designed to preserve product identity, purity, potency, and consistency.
When cellular starting material is collected using sterile, single‑use, closed or functionally closed systems, risk is driven far more by operator behaviour, process control, and system design than by air classification. Cleanrooms are essential when cells or reagents are openly manipulated; they are unnecessary when the product remains contained.
This model is not theoretical. In the US alone, tens of millions of blood and blood components are transfused safely each year, collected in controlled, non‑classified environments using these same principles. Ultimately, it is not the design of the facility but the discipline of the process and the integrity of the closed system that protect product quality.






