
Pharmaceutical manufacturers and regulators share a common goal to ensure a safe and effective finished product that is free from contamination. Part of the oversight process includes regulatory audits where personnel visit the facility to review for compliance with regulations.
While this process can be anxiety-inducing and nerve-wracking, a company that has embraced quality culture can view the experience as a positive check on their contamination control strategy, environmental monitoring, risk management, and data integrity.
The FDA warning letter describes the violation and the potential problem, followed by specifying what evidence the manufacturer must present to document the addressed concern
The US Food and Drug Administration (FDA) is one of the most transparent regulatory agencies as they publish warning letters of significant FDA violations by any manufacturer under their jurisdiction.
The FDA warning letter describes the violation and the potential problem, followed by specifying what evidence the manufacturer must present to document the addressed concern. This information is a valuable tool for companies to learn how to interpret and implement regulations and ensure a robust and appropriate quality culture. Thus, a review of FDA warning letters can be educational and insightful.
No post on Sundays
How did the COVID-19 pandemic impact FDA inspections and are there any trends that reflect industry changes? Just like every other company, the FDA faced challenges with staffing, difficulty in remote inspections, and pressures to release products to help patients. The FDA used a risk-based approach to determine the priority of domestic inspections due to the pandemic, which included some remote inspections.
From the FDA website, there were an average of 2,530 inspections each year for drug manufacturers pre-COVID and around 1,100 inspections each year for drug manufacturers post-COVID.
While the number of inspections decreased, was there a change in common citations? Thanks to the FDA’s transparency, top citations pre- and post-COVID can be viewed at FDA Dashboards - Inspections. While there were little changes in the three years prior to COVID to the three years during COVID, those changes highlight the unique trials and encounters of COVID-related supply chain shortages, staffing challenges, and pivots in production.
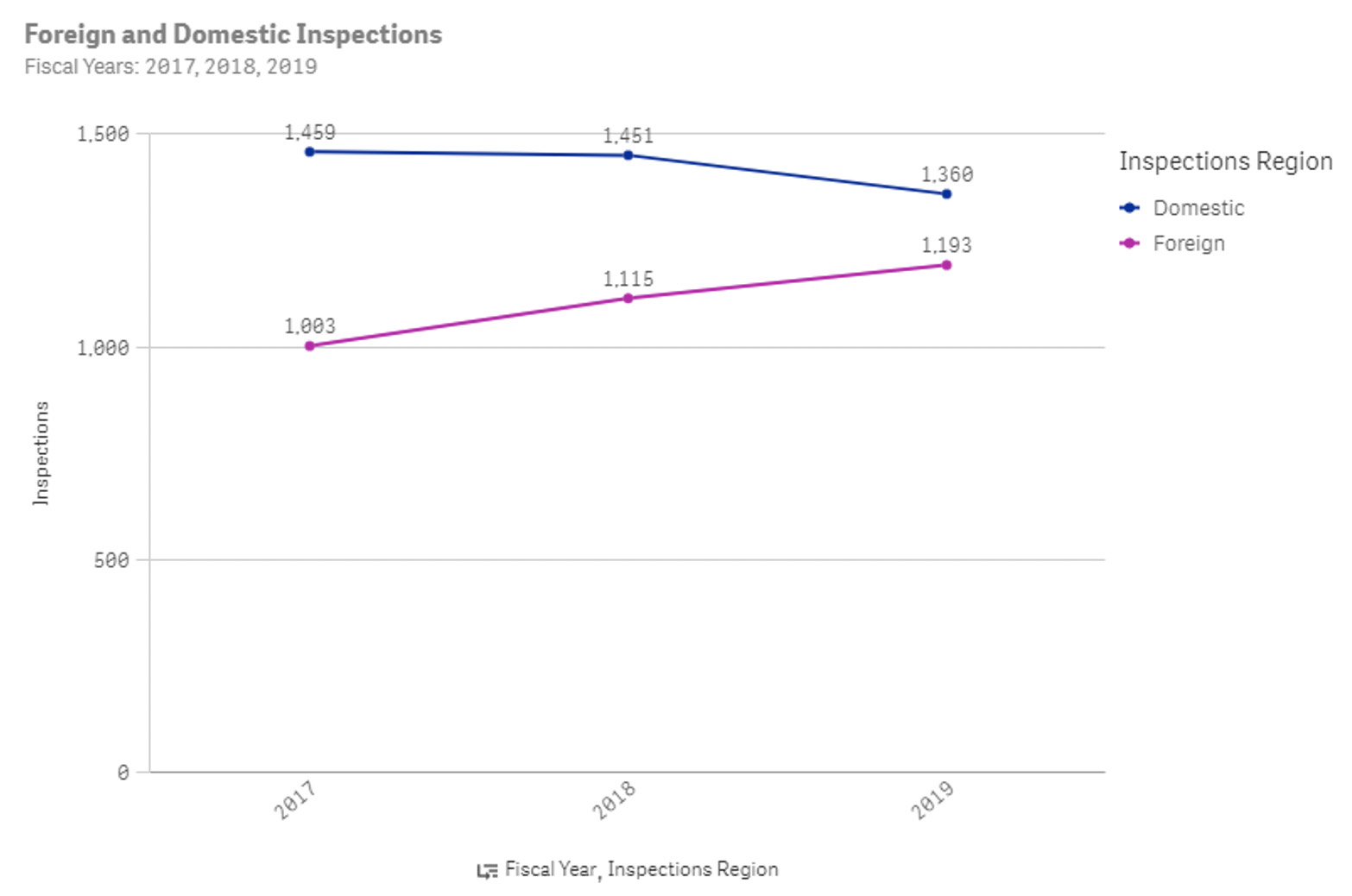
Figure 1: Number of FDA inspections for drug manufacturers in 2017, 2018, and 2019
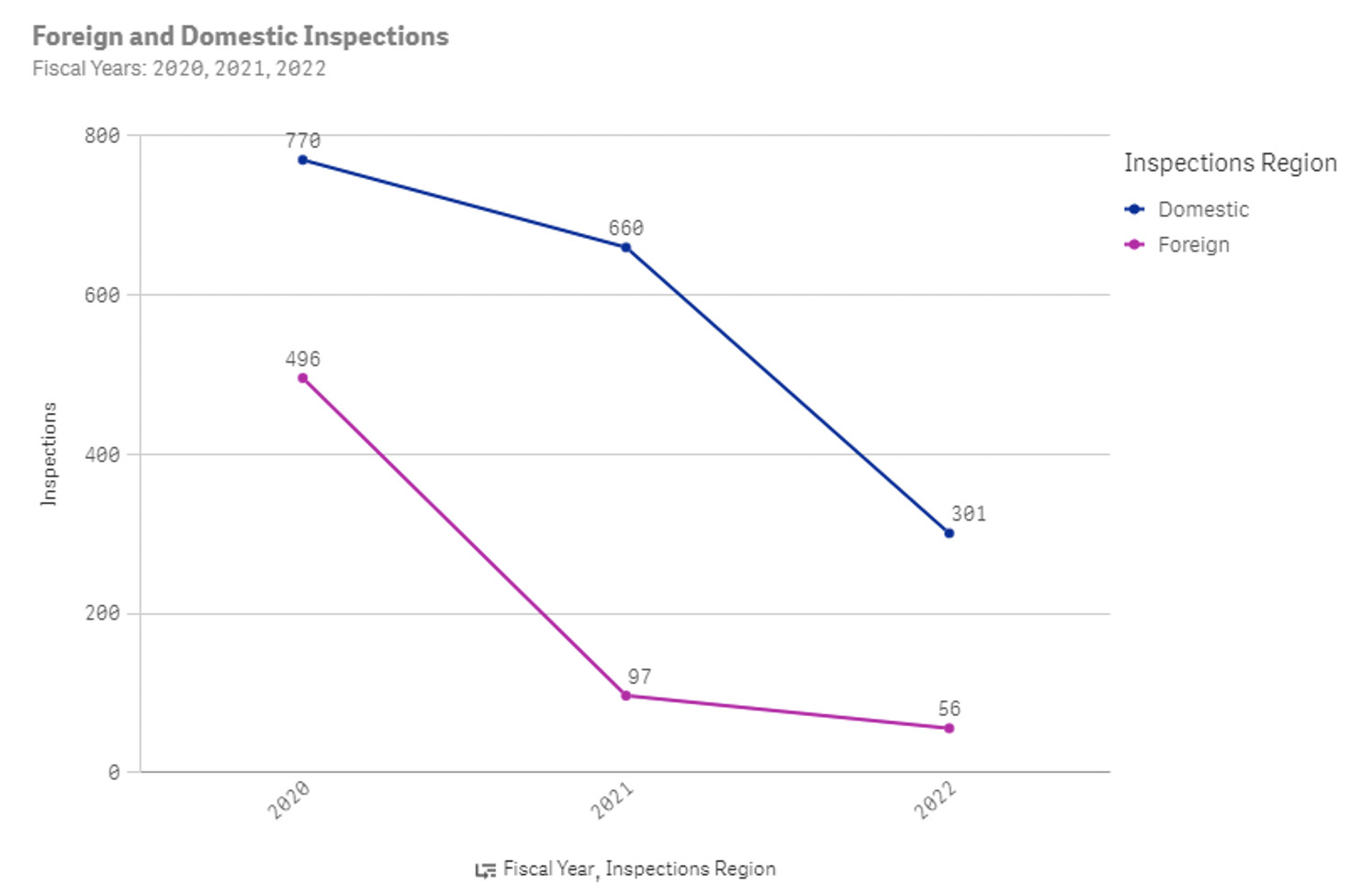
Figure 2: Number of FDA inspections for drug manufacturers in 2020, 2021, and 2022 YTD
How did citations change?
Citations for 21 CFR 211.192 (“Your firm failed to thoroughly investigate any unexplained discrepancy or failure of a batch or any of its components to meet any of its specifications, whether or not the batch has already been distributed”) increased from 11% of all citations to 13%. Pre-COVID, many of the citations were related to failed laboratory testing, lack of facility maintenance, and lack of root cause investigations.
These are still common topics as evidenced by recent warning letters: “Your water system was subject to microbiological contamination and numerous deviations from operating parameters since validation. Many of these deviations were either not investigated or did not result in implementation of CAPA. No investigations were conducted in response to identification of Burkholderia contaminans in seven samples taken from your water system ports (b)(4), and (b)(4), between January 1, 2020, and June 30, 2020. Burkholderia contaminans was not identified as an objectionable organism in your procedures or your (b)(4) water specifications. The presence of objectionable microorganisms in your (b)(4) water system can adversely affect the quality of your drug products”.
Citations for 21 CFR 211.67(a) (“Your firm failed to clean, maintain, and, as appropriate for the nature of the drug, sanitise and/or sterilise equipment and utensils at appropriate intervals to prevent malfunctions or contamination that would alter the safety, identity, strength, quality, or purity of the drug product beyond the official or other established requirements.”) also increased from 7% of all citations to 8%. Warning letter 622087 stated: “You did not validate the cleaning processes for the shared, product-contact equipment used to manufacture OTC drug products including equipment also used to manufacture non-drug products. Chemical and microbiological residues on equipment from previous manufacturing activities can adversely impact the purity, quality, and safety of drug products also manufactured on that equipment”.

Table 1: Top 10 FDA Citations for Drug Manufacturers in 2017, 2018, and 2019

Table 2: Top 10 FDA Citations for Drug Manufacturers in 2020, 2021, and 2022
New to the Top 10 citation list for drugs post-COVID is 21 CFR 211.63 (“Your firm failed to use equipment in the manufacture, processing, packing, or holding of drug products that is of appropriate design, adequate size, and suitably located to facilitate operations for its intended use and for its cleaning and maintenance”). One example of this violation is Case # 614278: “Ensuring your water system, used to manufacture your OTC drug products was suitable for use. The water system produced water contaminated with microorganisms recorded at a level of TNTC on March 23, 2020. Despite these findings, your QU did not exercise its authority to reject drug components made from this system and instead your firm continued to manufacture and release OTC drug products with objectionable microbiological contamination”.
These common citations make sense in light of the pandemic. The quick change in beginning manufacturing or changing manufacturing led to inadequate validation studies, investigations, or equipment. However, COVID-19 is not an excuse for noncompliance.
One last point related to FDA warning letters is the continued focus on data integrity. This trend has increased over the years and COVID-19 did not distract from it. In the FDA’s “Data Integrity and Compliance with Drug CGMP Questions and Answers, Guidance for Industry” the FDA clearly states that they “expect that all data be reliable and accurate”. They go on to emphasise that “ensuring data integrity is an important component of industry’s responsibility to ensure the safety, efficacy, and quality of drugs.”
Warning letter #586153 is a disturbing example of falsifying data. “Our inspection revealed serious data integrity breaches and other serious violations relating to environmental and personnel monitoring. We found that plates taken from ISO 5 areas exceeded action limits, but your firm failed to initiate investigations.
Citations for 21 CFR 211.192 increased from 11% of all citations to 13%
Furthermore, laboratory technicians falsified this data which is critical to maintaining an ongoing state of control in your aseptic processing facility. For instance, an environmental monitoring plate was recorded by your technician as “0” on your viable surface monitoring report form. The discarded plate was retrieved that same day and observed to contain colonies too numerous to count. In addition, although you failed to conduct “post activity” personnel monitoring for up to a year, your technicians repeatedly recorded results of “0” on the personnel (b)(4) report form. Personnel monitoring samples are critical because they indicate whether or not personnel in the aseptic processing environment are adversely affecting quality.
Due to this lack of authentic data relating to the microbial control of personnel who perform aseptic processing operations, you lacked information that is basic to determining aseptic processing control. For up to a year, you lacked the ability to identify microbial contamination risks posed by personnel.” While such blatant disregard of integrity is unusual, this letter highlights the obligation to continuous improvement and contamination control.
The COVID-19 pandemic posed an incredible number of problems, but those problems never justify a disregard for quality and safety. COVID-19 put an intense focus on pharmaceutical manufacturing. Patients were waiting for treatments and relying on those companies for product safety. This should be a shared mission between regulators and manufacturers, which requires communication and continued improvement. Learning from FDA warning letters is just one way demonstrate a commitment to quality, safety, and excellence.



